Recently, a new study describes an evolutionary tree that quickly obtains precise dates — also known as the "tree of time" – a new method of calculation. With this new approach, the researchers analyzed a dataset of mammalian genomes and answered the long-standing question of whether modern fetal mammal populations originated before or after the Cretaceous-Archean (K-Pg) mass extinction.
That mass extinction wiped out more than 70 percent of species, including all dinosaurs.
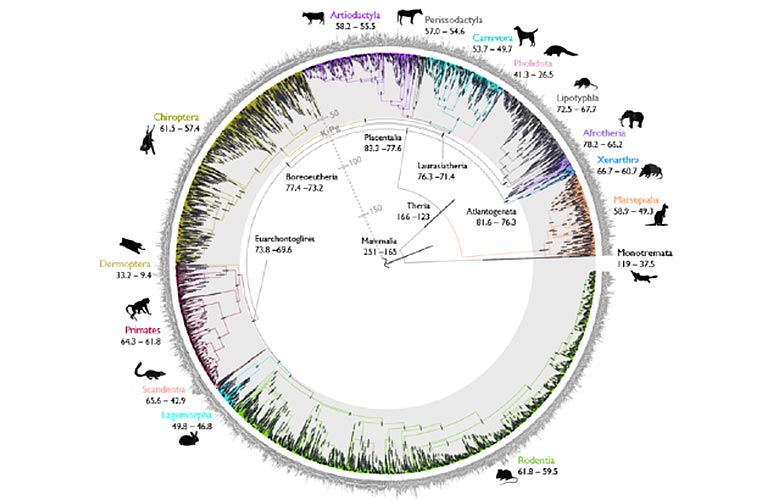
These findings confirm that the ancestors of modern placental mammal populations followed the K-Pg mass extinction that occurred 66 million years ago, which resolves the controversy surrounding the origins of modern mammals. Placental mammals are the most diverse group of living mammals, including primates, rodents, cetaceans, carnivores, pterodactyls (bats), and humans.
The research team was led by Dr Mario dos Reis (Queen Mary University of London) and Professor Phil Donoghue (University of Bristol) and included scientists from Queen Mary' College, the University of Bristol, the University of London, Imperial College London and the University of Cambridge.
Dr Sandra Álvarez-Carretero, lead author of the paper from UCL (then at Queen Mary University), said: "By integrating the complete genome and necessary fossil information in the analysis, we were able to reduce uncertainty and obtain an exact evolutionary timeline. Did modern mammals coexist with dinosaurs or did they originate after the mass extinction? We now have a definitive answer. ”
"The timeline of mammalian evolution is perhaps one of the most controversial topics in evolutionary biology. Early studies have provided estimates of the origins of modern protopes in the deep Cretaceous, dinosaur era. Over the past 20 years, research has shuttled back and forth between the diversification schemes after K-Pg and before K-Pg," added Professor Donogue, co-first author of the paper. ”
A rapid method of genomic analysis
With sequencing projects around the world now producing hundreds of genome sequences and about to plan to sequence more than a million species, evolutionary biologists will soon have a wealth of information in their hands. However, current methods of analyzing existing large genomic datasets and creating evolutionary timelines are inefficient and computationally expensive.
"Extrapolating the timeline of evolution is a fundamental goal of biology. However, state-of-the-art methods rely on using computers to simulate evolutionary timelines and assess the most plausible timelines. In our case, it was difficult due to the analysis of huge data sets involving genetic data from nearly 5,000 species of mammals and 72 complete genomes," said Dr. dos Reis.
In this study, the researchers developed a new, rapid Bayesian method to analyze large numbers of genomic sequences, while also taking into account uncertainties in the data. "We addressed the computational barrier by dividing the analysis into sub-steps: first using 72 genome simulation timelines, and then using the results to guide the simulation of the rest of the species," dr. dos Reis noted, "using the genome reduces uncertainty because it can reject unreliable timelines from simulations." ”
"Our data processing pipeline captures as much genomic data as possible for as many mammalian species as possible. This is challenging because genetic databases contain inaccurate content and we have to develop a strategy to identify poor quality samples or mislabeled data that must be removed," added Dr. Asif Tamuri, co-first author of the paper from UCL. He is responsible for assembling the mammalian genome dataset.
More efficient and sustainable
With this new approach, the team was able to reduce the computational time for this complex analysis from decades to months. "If we hadn't tried to analyze this large mammal dataset in a supercomputer using the Bayesian method we developed, we would have had to wait decades to infer the mammalian time tree," Álvarez-Carretero said. In addition, we managed to reduce the calculation time by a factor of 100. This new method not only analyzes genomic datasets, but also significantly reduces the amount of carbon dioxide released as a result of the calculations due to its higher efficiency. ”
The methods developed in the study could be used to address other controversial evolutionary timelines that require analysis of large data sets. By combining the novel Bayesian method with the genomes of the upcoming Darwin Tree of Life and Earth BioGenome projects, the idea of estimating a reliable evolutionary timescale for the tree of life now seems possible.
![Name: Coordinated Work Alias: Harmony English Name: Harmony Creator: Alexander Cabanel Date: 1877 Category: Situational Material: Oil on Canvas[fig]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACwAAAAAAQABAAACAkQBADs=)