今天伯小远将带着大家一起通过将参考论文与演示实验结合起来的形式,探讨蛋白质-蛋白质互作与软件对接的问题,希望对大家研究蛋白质互作有所帮助。
技术背景
2005年,Wigge等人在Science上报道了拟南芥“诱导开花”的信号调控通路,简言之:在拟南芥体内有一个FLOWERING LOCUS T(FT)基因,其编码的蛋白可与一个bZIP族的转录因子FD形成复合体,从而激活APETALA1(AP1)的表达,其中AP1是诱导开花的关键蛋白,并且,他们推测该通路在高等植物中可能是保守的(Wigge et al., 2005)。
水稻中有一个Hd3a的蛋白,与拟南芥FT有很高的同源性。Taoka等人发现其调控机制可能与拟南芥FT-FD-AP1通路类似,因此展开了一系列研究(Taoka et al., 2011)。研究的结论,咱们从以下三个实验结果就可以一目了然了。
实验一:
OsMADS15与拟南芥AP1是高度同源的蛋白,是水稻中诱导开花的关键蛋白。OsFD1与拟南芥FD是功能相似的蛋白,可能是OsMADS15的转录因子。如下图,进行转录水平分析时发现,单独存在Hd3a和OsFD1都无法激活OsMADS15的表达,只有两者共存时才可以。
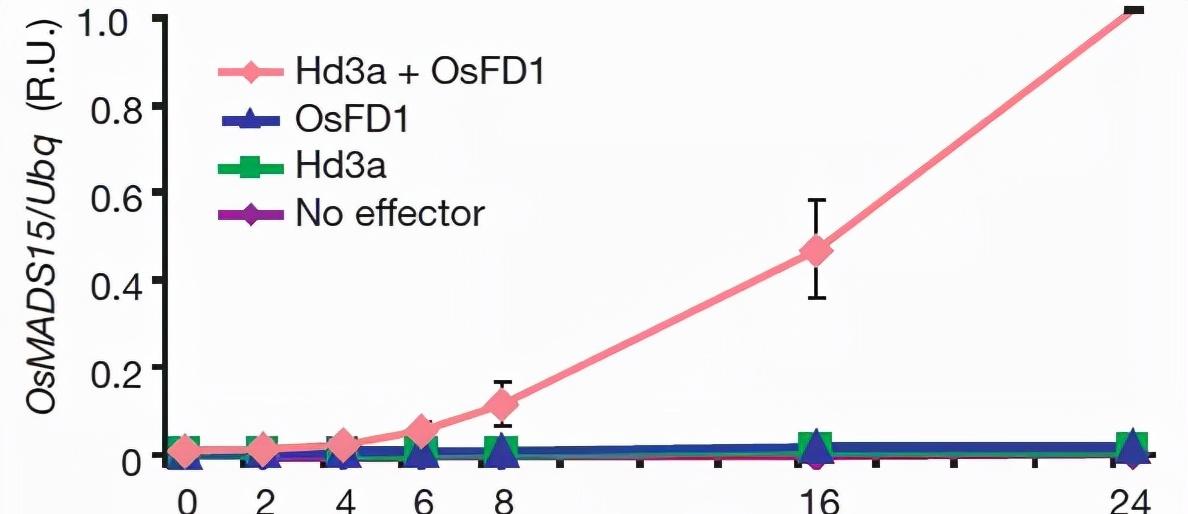
图1 Hd3a和OsFD1调控OsMADS15表达水平。
实验二:
GST pull-down结果表明(图2),Hd3a与GF14c(也叫14-3-3c)互作,GF14c与OsFD1互作,Hd3a与OsFD1不互作,但三者同时存在时也互作。因此,推论Hd3a对OsFD1的互作需要GF14c介导。

图2 GST pull-down验证Hd3a、GF14c(14-3-3c)、OsFD1三者之间的互作。
实验三:
EMSA实验表明(图3左),OsFD1可以与OsMADS15的启动子结合,但只有与Hd3a、GF14c形成复合结构,才能实现对OsMADS15的转录激活。综上,水稻开花调控途径为:Hd3a在叶片细胞表达,经运输到达茎尖细胞,14-3-3家族蛋白作为胞内受体,与Hd3a结合形成复合体,并在OsFD1的作用下进入细胞核,与OsFD1形成三元复合体(图3右),三元复合体可以激活OsMADS15的表达,从而完成对开花的诱导。
图3 EMSA结果和推测的Hd3a--14-3-3c--OsFD1的DNA复合结构。
蛋白质-蛋白质对接
通过上面的文献解析,发现论文的重点就是研究Hd3a、14-3-3c、OsFD1三类蛋白间的相互作用。如果把互作蛋白对比作“情侣”,那么故事就是:Hd3a、14-3-3c相爱,14-3-3c、OsFD1相爱,但Hd3a、OsFD1不相爱。因为“爱”(互作),三者放下偏见,协调起来共同完成了促进“开花”这一结果。而我们的任务就是,利用蛋白质-蛋白质对接软件,从理论模拟上重现这场“爱情故事”。先说明一下,Hd3a/14-3-3c的复合结构,已经被Taoka等解析了,单位了给大家讲解,我们可以假装他们的结构还未解析,然后以拟南芥(或烟草)的同源蛋白为模板,构建三维结构,再通过对接软件模拟互作过程。
在对接之前,首先要分析下任务。从论文报道的二元复合结构的剖视图来看(图4),可能发生互作的有Hd3a与 14-3-3c、14-3-3c与14-3-3c、14-3-3c与OsFD1、OsFD1与OsFD1,但是OsFD1的结构实在难以获得,因此对于它的互作不做分析。
图4 Hd3a、14-3-3c、OsFD1复合结构的3D图。
1. 准备好两个蛋白的PDB文件
对于如何获得PDB文件,伯小远在这里先给大家介绍两个常用的方法:
(1)如果目标蛋白的结构在RSCP PDB数据库中已经存在,则可以直接下载。如果不存在,则可以通过同源建模和从头预测软件构建目标蛋白的三维结构。
(2)今年7月份报道的AlphaFold2在蛋白质结构预测方面表现卓越,已获得人、水稻、拟南芥、玉米等二十多个物种的全部预测蛋白,可以在UniPort等数据库上直接下载。
如果是RSCP PDB数据库里没有,AlphaFold2中也没有预测的蛋白质,该怎么办呢?别急,伯小远在这里给大家介绍一个在线同源建模的方法——SWISS-MODEL(网址https://swissmodel.expasy.org/),该方法不需要安装任何软件,并且,如果同源建模的模板与目标蛋白的序列一致性较高(具体多高算高,其实没有一个统一的定论,大于60%已经可以得到高质量的三维模型),那么同源建模的准确性就会极高,甚至优于AlphaFold2预测。
SWISS-MODEL是使用最为广泛的同源建模在线软件,没有之一,而且免费哟!其操作简单,只需填入序列、项目名称等信息,即可开始同源建模。
图5 SWISS-MODEL序列提交页面。
程序运行结束,可以在“Model Results”界面查看建模结果(图6)。其中三个关键参数分别为:
(1)六边形里的代表模型的质量,数值越接近1越好。
(2)椭圆形里的代表与模板序列的一致性,数值越接近100%越好。特别注意的是,要看一下“Coverage”,它代表模板对目标蛋白的覆盖度,程序只用已有结构的部分建模。
(3)长方形里的代表建模结果,一般选择PDB格式。
图6 SWISS-MODEL运行结果页面。
注:有时从RSCP PDB数据库中下载的文件会存在氨基酸信息缺失现象,从而导致后续的分析程序报错,需要先用SPDBV软件打开,如果含有缺失序列,软件会自动补全;同源结构建模或者AlphaFold2、RoseTTAFold等软件构建的三维结构模型不存在这个问题。
表1 待模拟结构与参考结构比对结果
a:虽然AlphaFold2已把水稻和拟南芥所有的蛋白结构已经预测出来了,但OsFD1/AtFD比较特殊,它们含有大量不规则卷曲片段(Loop区),一般在Loop长度超过12个氨基酸的情况下,如果没有可靠的模板或限制信息,几乎无法通过预测构建结构。而OsFD1/AtFD的不规则区有100多个氨基酸,AlphaFold2也没有办法。好在OsFD1的A123-V177区域是一段保守的α螺旋,也是bZIP类蛋白的共有结构域,因此可截取123-195区间的肽段来进行预测结果。
根据上表的信息,我们通过同源建模,逐一建立Hd3a、14-3-3c、OsFD1的三维结构。由于Hd3a、14-3-3c已有晶体结构,如果直接运行同源建模,程序会以自己为模板模拟自己(图7)。因此,我们在建模开始界面先执行“Search For templates”,然后选择表中的参考模板进行建模。
图7 同源建模运行界面(以Hd3a为例)。
完成建模后,可以通过在线工具SuperPose(网址:http://superpose.wishartlab.com/)对比模型(以Hd3a为例)的准确性,从下图可以看出,相较于Hd3a的晶体结构,同源建模与AlphaFold2的准确性相当,在全原子层面,同源建模甚至更胜一筹(RMSD越小,两个三维结构越接近,一般全部原子的RMSD小于2,认为两个结构已经十分接近了)。
图8 同源建模和从头预测结构与真实结构的比较。
2. 分子对接
蛋白结构准备完成后,就可以进入对接步骤啦!由于蛋白-蛋白对接是一个十分复杂的事情,影响因素有很多,预测算法也从一开始的基于FFTs算法的刚性对接(ZDOCK),发展到现在整合多步骤的HADDOCK、ClusPro、SwamDock等。尤其是近年来,随着机器学习算法的发展以及实验数据的积累,一些基于共进化、同源蛋白的互作预测算法,显著提高了蛋白质-蛋白质预测的准确性。下面,伯小远就来为大家介绍两条技术路线:
(1)基于ZDOCK-RosettaDock的方法
ZDOCK-RosettaDock方法是先将两个大分子进行基于全局算法的刚性对接,就好比将一对情侣拉到“当面”聊一聊。其过程是一个分子不动,另一个分子从各种位置靠近它,最终选出得分最高的结构。不过“百炼钢终敌不过绕指柔”,对接模式就好比“择偶标准”,也是因人而异的,当一个分子接近另一个分子时,其表面构象是会发生变化的,因此,就有了RosettaDock局部精细对接,它允许对接分子进行构象选择和调整。
ZDOCK-RosettaDock方法既有本地版也有在线版,但本地版对于非专业人员上手难度较大,本文不做详细介绍,下面仅介绍在线版的使用方法:
1)ZDOCK
ZDOCK需要提供学校邮箱才能使用,具体参数设置可参考。下载好结果后,需检查PDB文件,确保每个结构末尾均添加了“TER”字符,否则RosettaDock步骤可能会出错。
2)RosettaDock
RosettaDock在线服务器(Lyskov et al., 2008)提供了Rosetta程序集的主要功能,其中“[Docking2]”代表RosettaDock模块。网站自带使用说明,只需提供单体结构,填写肽链信息等,使用十分简便哟!
上面的方法对接效果如何呢?伯小远首先从已报道的Hd3a/14-3-3c复合结构中,利用SPDBV软件把Hd3a/14-3-3c的异二聚体结构提取出来,然后把对接得到的复合结构与实验数据进行结构拟合,结果如下图所示。
图9 ZDOCK-RosettaDock的对接结果与真实结构的比较。
脸打的啪啪响啊,预测与事实根本不一致(图9中14-3-3c是重叠的,但真实的Hd3a与预测的Hd3a一左一右,方位完全不同)。怎么办呢?别怕,咱们还有套路。
(2)基于限制信息的HADDOCK方法
上面的对接结果之所以不准确,是因为对接分子是从完全随机的位置开始的,而且,因为ZDOCK是刚性对接软件,评分时偏重于互作界面面积较大的方位,这与事实可能是不符的。如果能够获得两个对接分子的初始位置,或者位于互作界面的氨基酸信息,那么对结果的准确性会显著提高。伯小远介绍以下两种情况:
1)有限制性残基的实验数据 如文章中的酵母双杂结果表明,Hd3a的R64、P96、F103、R132以及14-3-3c的F200、I204、Y215都可能是互作界面的关键残基,因为它们突变后,双杂结果由阳性变为阴性。如果把它们在晶体结构上标出来,就可以很直观的看到了(图10)。这些界面上的关键残基,在蛋白质对接中叫做限制性残基,若能够通过实验获取它们的信息,那么利用HADDOCK进行对接,准确性极高。
图10 限制性残基的位置。
2)有同源复合结构做参考
如果已经报道了同源蛋白的结构,或者结构功能相似的同类复合结构,则可以通过结构拟合先把两个待对接的单体分子与参考复合结构叠加,然后撤掉参考结构。这时两个对接分子的空间位置,就是一个很好的起始位置,以此进行ZDOCK-RosettaDock对接,或者推测出限制性残基信息进行HADDOCK对接,都能够得到比较精确的预测结果。
HADDOCK(https://wenmr.science.uu.nl/haddock2.4/)是一款非常优秀的软件,使用前需注册。该软件需提供两个待对接分子以及限制性残基的相关信息,伯小远在加入Hd3a和14-3-3c的限制残基信息后,获得的模拟二聚体与真实的晶体结构重叠性极好(这里图片不再展示)。另外,该软件允许修改大量的参数,灵活性极高,输出内容丰富而精美,软件也可用于实际研究。(网站上提供了使用说明,对蛋白质-蛋白质对接有兴趣或者有需求的同学,值得好好研究一番)。
3. 对接结果的互作分析
两个蛋白质能够相互结合,主要取决于它们之间的静电引力和范德华力,具体包括:互作界面的电荷分布、几何形状互补面积、氢键、盐桥、疏水相互作用、芳环堆积作用等。蛋白质对接的物理、化学原理,模型评价方法是十分复杂的,本文先不讨论。伯小远仅针对对接结果,提出一般的评估方法。
重点推荐PDBePISA(https://www.ebi.ac.uk/msd-srv/prot_int/pistart.html),该软件在分析大分子溶剂可及性和相互作用界面方面非常优秀,其可通过输入复合结构的RSCP PDB编号或上传对接的复合结构来进行分析,使用方法十分简单,但面对预测结果,又该如何理解呢?
首先,信息汇总栏的各项重点指标已在下图标出,有些参数越大越好,有些越小越好。但没有明确的能判断是否互作的标准,因此,最好采用同源蛋白或同类蛋白复合物的结构做为参考;其次,氢键、盐桥的数目越多越好,距离越短越好,也没有参考阈值;最后,PDBePISA有一个参数叫复合结构显著性分数(CSS),它并不是指示互作可信度的,而是指该界面在复合体形成中的重要程度,即使CSS=0,也不能代表两分子间没有互作,但若CSS>0,则两分子间极可能存在互作。
图11 PDBePISA对Hd3a/14-3-3c互作界面的分析结果。
三元复合体的组装
上面一系列的操作获得了Hd3a/14-3-3c的二元结构(图片未展示),而三元结构是两个Hd3a/14-3-3c二元结构依靠14-3-3c和14-3-3c互作联系起来的,因此还需要构建14-3-3c的同二聚体结构。14-3-3c同二聚体没有限制性残基信息,但其建模用的高同源模板(烟草14-3-3c),有同二聚体晶体结构,因此可利用该同源复合结构进行起始位置的初猜,经过ZDOCK分析,模拟的14-3-3c同二聚体复合结构与报道也基本一致(图片未展示)。
然后,就是Hd3a/14-3-3c、14-3-3c/14-3-3c两个二元结构的叠加,用SPDBV载入两个结构,选中全部原子,执行“Fit”菜单的“Magic Fit”,可以快速进行结构叠加(或者叫拟合),经过反复拟合结果展示如下(图12),与文章报道几乎一模一样。
图12 模拟获得的Hd3a/14-3-3c复合结构。
接下来,到了最关键的OsFD1的叠加。这一步非常困难,由于OsFD1的结构是不准的,报道中的晶体仅包含了OsFD1 189-195的7个残基,也无法与AlphaFold2的预测结果拟合,叠加的方法完全行不通。因此,只能依靠酵母双杂的限制性信息,使用HADDOCK强行对接。最终全部叠加的结果如下图,Hd3a/14-3-3c部分与真实结构基本一致,OsFD1部分与文中的推测有较大差异。注:Taoka等也是靠推测得到的结果,他们的OsFD1是用小鼠的同类蛋白叠加的,可能也不是真实结构哟。
图13 分子对接和叠加获得的最终结构。
至此,“剧情”已经不能完全按“编剧”的设想发展了,这场“爱情故事”到此结束!
仅仅依靠模拟就走到这种程度已经非常不容易了,当然需要承认的是,伯小远在这里所选的目标蛋白都有不错的高同源模板,并且有相关的实验结果做指导。如果没有这些,预测的方法是有很大局限性的。为便于大家尝试,本文提供的方法大多是在线版本的,若需用于研究,建议一定要采用本地版,灵活调整参数,不断筛选、重复,并配合一定实验验证,才能取得更为可靠的结果。
小 结
最后,请大家思考一个问题,为什么要做那么多不同的实验来验证蛋白间的互作呢?估计有的同学会回答,可以让论文内容看起来更丰富,容易发表。我只能说,你真是个小机灵鬼!
如果认真看过我们公众号以前的文章,可能还记得,我们曾比较过不同的蛋白互作验证技术间的优缺点,如果只通过一、两种验证方式,可能无法了解事实的真相,也可能得到相悖的结果,这是完全正常的。在Taoka等的研究中,也出现了酵母双杂的结果与GST pull-down及核磁共振化学位移结果不一致的情况,这主要是由于OsFD1的192号氨基酸磷酸化导致的,GST pull-down及核磁共振是使用原核表达的高纯蛋白,没有磷酸化修饰,而酵母内是有磷酸化修饰的。但作者同时采用BiFC对其结果进行验证,表明OsFD1与Hd3a可能存在互作关系。
酵母双杂、BiFC、FERT 、Co-IP、GST pull-down、核磁共振,包括本文未涉及的其他技术,如蛋白-蛋白相互作用陷阱、生物膜干涉等技术,都会因胞内、胞外,有无蛋白质修饰,亚细胞定位是否相同,是否有第三方参与,互作强弱等问题产生差异,解决的办法就是设置严谨的对照、采用尽可能多的方法进行验证。至于已经忘得一干二净的同学,非常有必要再回顾一下《基因功能研究的那些套路你知道多少?(下)》。
References:
protein docking. Nucleic Acids Res. 2008, 36(Web Server issue):W233-8.
Purwestri YA, Ogaki Y, Tamaki S, et al. The 14-3-3 protein GF14c acts as a negative regulator of flowering in rice by interacting with the florigen Hd3a. Plant Cell Physiol. 2009, 50(3):429-38.
Taoka K, Ohki I, Tsuji H, et al. 14-3-3 proteins act as intracellular receptors for rice Hd3a florigen. Nature. 2011, 476(7360): 332-5.
Wigge PA, Kim MC, Jaeger KE, et al. Integration of spatial and temporal information during floral induction in Arabidopsis. Science. 2005, 309(5737): 1056-9.