今天伯小遠将帶着大家一起通過将參考論文與示範實驗結合起來的形式,探讨蛋白質-蛋白質互作與軟體對接的問題,希望對大家研究蛋白質互作有所幫助。
技術背景
2005年,Wigge等人在Science上報道了拟南芥“誘導開花”的信号調控通路,簡言之:在拟南芥體内有一個FLOWERING LOCUS T(FT)基因,其編碼的蛋白可與一個bZIP族的轉錄因子FD形成複合體,進而激活APETALA1(AP1)的表達,其中AP1是誘導開花的關鍵蛋白,并且,他們推測該通路在高等植物中可能是保守的(Wigge et al., 2005)。
水稻中有一個Hd3a的蛋白,與拟南芥FT有很高的同源性。Taoka等人發現其調控機制可能與拟南芥FT-FD-AP1通路類似,是以展開了一系列研究(Taoka et al., 2011)。研究的結論,咱們從以下三個實驗結果就可以一目了然了。
實驗一:
OsMADS15與拟南芥AP1是高度同源的蛋白,是水稻中誘導開花的關鍵蛋白。OsFD1與拟南芥FD是功能相似的蛋白,可能是OsMADS15的轉錄因子。如下圖,進行轉錄水準分析時發現,單獨存在Hd3a和OsFD1都無法激活OsMADS15的表達,隻有兩者共存時才可以。
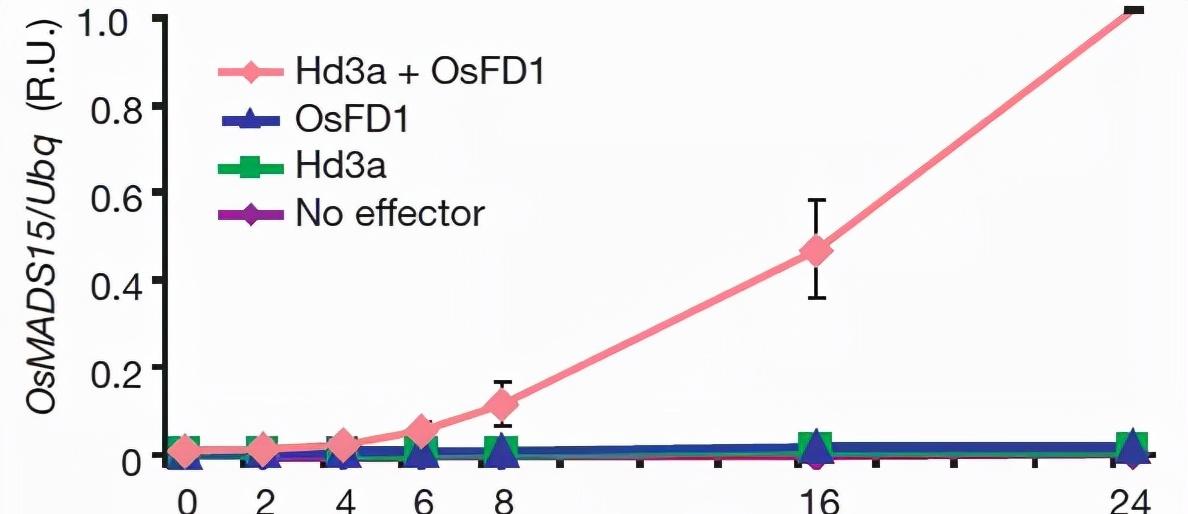
圖1 Hd3a和OsFD1調控OsMADS15表達水準。
實驗二:
GST pull-down結果表明(圖2),Hd3a與GF14c(也叫14-3-3c)互作,GF14c與OsFD1互作,Hd3a與OsFD1不互作,但三者同時存在時也互作。是以,推論Hd3a對OsFD1的互作需要GF14c介導。

圖2 GST pull-down驗證Hd3a、GF14c(14-3-3c)、OsFD1三者之間的互作。
實驗三:
EMSA實驗表明(圖3左),OsFD1可以與OsMADS15的啟動子結合,但隻有與Hd3a、GF14c形成複合結構,才能實作對OsMADS15的轉錄激活。綜上,水稻開花調控途徑為:Hd3a在葉片細胞表達,經運輸到達莖尖細胞,14-3-3家族蛋白作為胞内受體,與Hd3a結合形成複合體,并在OsFD1的作用下進入細胞核,與OsFD1形成三元複合體(圖3右),三元複合體可以激活OsMADS15的表達,進而完成對開花的誘導。
圖3 EMSA結果和推測的Hd3a--14-3-3c--OsFD1的DNA複合結構。
蛋白質-蛋白質對接
通過上面的文獻解析,發現論文的重點就是研究Hd3a、14-3-3c、OsFD1三類蛋白間的互相作用。如果把互作蛋白對比作“情侶”,那麼故事就是:Hd3a、14-3-3c相愛,14-3-3c、OsFD1相愛,但Hd3a、OsFD1不相愛。因為“愛”(互作),三者放下偏見,協調起來共同完成了促進“開花”這一結果。而我們的任務就是,利用蛋白質-蛋白質對接軟體,從理論模拟上重制這場“愛情故事”。先說明一下,Hd3a/14-3-3c的複合結構,已經被Taoka等解析了,機關了給大家講解,我們可以假裝他們的結構還未解析,然後以拟南芥(或煙草)的同源蛋白為模闆,建構三維結構,再通過對接軟體模拟互作過程。
在對接之前,首先要分析下任務。從論文報道的二進制複合結構的剖視圖來看(圖4),可能發生互作的有Hd3a與 14-3-3c、14-3-3c與14-3-3c、14-3-3c與OsFD1、OsFD1與OsFD1,但是OsFD1的結構實在難以獲得,是以對于它的互作不做分析。
圖4 Hd3a、14-3-3c、OsFD1複合結構的3D圖。
1. 準備好兩個蛋白的PDB檔案
對于如何獲得PDB檔案,伯小遠在這裡先給大家介紹兩個常用的方法:
(1)如果目标蛋白的結構在RSCP PDB資料庫中已經存在,則可以直接下載下傳。如果不存在,則可以通過同源模組化和從頭預測軟體建構目标蛋白的三維結構。
(2)今年7月份報道的AlphaFold2在蛋白質結構預測方面表現卓越,已獲得人、水稻、拟南芥、玉米等二十多個物種的全部預測蛋白,可以在UniPort等資料庫上直接下載下傳。
如果是RSCP PDB資料庫裡沒有,AlphaFold2中也沒有預測的蛋白質,該怎麼辦呢?别急,伯小遠在這裡給大家介紹一個線上同源模組化的方法——SWISS-MODEL(網址https://swissmodel.expasy.org/),該方法不需要安裝任何軟體,并且,如果同源模組化的模闆與目标蛋白的序列一緻性較高(具體多高算高,其實沒有一個統一的定論,大于60%已經可以得到高品質的三維模型),那麼同源模組化的準确性就會極高,甚至優于AlphaFold2預測。
SWISS-MODEL是使用最為廣泛的同源模組化線上軟體,沒有之一,而且免費喲!其操作簡單,隻需填入序列、項目名稱等資訊,即可開始同源模組化。
圖5 SWISS-MODEL序列送出頁面。
程式運作結束,可以在“Model Results”界面檢視模組化結果(圖6)。其中三個關鍵參數分别為:
(1)六邊形裡的代表模型的品質,數值越接近1越好。
(2)橢圓形裡的代表與模闆序列的一緻性,數值越接近100%越好。特别注意的是,要看一下“Coverage”,它代表模闆對目标蛋白的覆寫度,程式隻用已有結構的部分模組化。
(3)長方形裡的代表模組化結果,一般選擇PDB格式。
圖6 SWISS-MODEL運作結果頁面。
注:有時從RSCP PDB資料庫中下載下傳的檔案會存在氨基酸資訊缺失現象,進而導緻後續的分析程式報錯,需要先用SPDBV軟體打開,如果含有缺失序列,軟體會自動補全;同源結構模組化或者AlphaFold2、RoseTTAFold等軟體建構的三維結構模型不存在這個問題。
表1 待模拟結構與參考結構比對結果
a:雖然AlphaFold2已把水稻和拟南芥所有的蛋白結構已經預測出來了,但OsFD1/AtFD比較特殊,它們含有大量不規則卷曲片段(Loop區),一般在Loop長度超過12個氨基酸的情況下,如果沒有可靠的模闆或限制資訊,幾乎無法通過預測建構結構。而OsFD1/AtFD的不規則區有100多個氨基酸,AlphaFold2也沒有辦法。好在OsFD1的A123-V177區域是一段保守的α螺旋,也是bZIP類蛋白的共有結構域,是以可截取123-195區間的肽段來進行預測結果。
根據上表的資訊,我們通過同源模組化,逐一建立Hd3a、14-3-3c、OsFD1的三維結構。由于Hd3a、14-3-3c已有晶體結構,如果直接運作同源模組化,程式會以自己為模闆模拟自己(圖7)。是以,我們在模組化開始界面先執行“Search For templates”,然後選擇表中的參考模闆進行模組化。
圖7 同源模組化運作界面(以Hd3a為例)。
完成模組化後,可以通過線上工具SuperPose(網址:http://superpose.wishartlab.com/)對比模型(以Hd3a為例)的準确性,從下圖可以看出,相較于Hd3a的晶體結構,同源模組化與AlphaFold2的準确性相當,在全原子層面,同源模組化甚至更勝一籌(RMSD越小,兩個三維結構越接近,一般全部原子的RMSD小于2,認為兩個結構已經十分接近了)。
圖8 同源模組化和從頭預測結構與真實結構的比較。
2. 分子對接
蛋白結構準備完成後,就可以進入對接步驟啦!由于蛋白-蛋白對接是一個十分複雜的事情,影響因素有很多,預測算法也從一開始的基于FFTs算法的剛性對接(ZDOCK),發展到現在整合多步驟的HADDOCK、ClusPro、SwamDock等。尤其是近年來,随着機器學習算法的發展以及實驗資料的積累,一些基于共進化、同源蛋白的互作預測算法,顯著提高了蛋白質-蛋白質預測的準确性。下面,伯小遠就來為大家介紹兩條技術路線:
(1)基于ZDOCK-RosettaDock的方法
ZDOCK-RosettaDock方法是先将兩個大分子進行基于全局算法的剛性對接,就好比将一對情侶拉到“當面”聊一聊。其過程是一個分子不動,另一個分子從各種位置靠近它,最終選出得分最高的結構。不過“百煉鋼終敵不過繞指柔”,對接模式就好比“擇偶标準”,也是因人而異的,當一個分子接近另一個分子時,其表面構象是會發生變化的,是以,就有了RosettaDock局部精細對接,它允許對接分子進行構象選擇和調整。
ZDOCK-RosettaDock方法既有本地版也有線上版,但本地版對于非專業人員上手難度較大,本文不做詳細介紹,下面僅介紹線上版的使用方法:
1)ZDOCK
ZDOCK需要提供學校郵箱才能使用,具體參數設定可參考。下載下傳好結果後,需檢查PDB檔案,確定每個結構末尾均添加了“TER”字元,否則RosettaDock步驟可能會出錯。
2)RosettaDock
RosettaDock線上伺服器(Lyskov et al., 2008)提供了Rosetta程式集的主要功能,其中“[Docking2]”代表RosettaDock子產品。網站自帶使用說明,隻需提供單體結構,填寫肽鍊資訊等,使用十分簡便喲!
上面的方法對接效果如何呢?伯小遠首先從已報道的Hd3a/14-3-3c複合結構中,利用SPDBV軟體把Hd3a/14-3-3c的異二聚體結構提取出來,然後把對接得到的複合結構與實驗資料進行結構拟合,結果如下圖所示。
圖9 ZDOCK-RosettaDock的對接結果與真實結構的比較。
臉打的啪啪響啊,預測與事實根本不一緻(圖9中14-3-3c是重疊的,但真實的Hd3a與預測的Hd3a一左一右,方位完全不同)。怎麼辦呢?别怕,咱們還有套路。
(2)基于限制資訊的HADDOCK方法
上面的對接結果之是以不準确,是因為對接分子是從完全随機的位置開始的,而且,因為ZDOCK是剛性對接軟體,評分時偏重于互作界面面積較大的方位,這與事實可能是不符的。如果能夠獲得兩個對接分子的初始位置,或者位于互作界面的氨基酸資訊,那麼對結果的準确性會顯著提高。伯小遠介紹以下兩種情況:
1)有限制性殘基的實驗資料 如文章中的酵母雙雜結果表明,Hd3a的R64、P96、F103、R132以及14-3-3c的F200、I204、Y215都可能是互作界面的關鍵殘基,因為它們突變後,雙雜結果由陽性變為陰性。如果把它們在晶體結構上标出來,就可以很直覺的看到了(圖10)。這些界面上的關鍵殘基,在蛋白質對接中叫做限制性殘基,若能夠通過實驗擷取它們的資訊,那麼利用HADDOCK進行對接,準确性極高。
圖10 限制性殘基的位置。
2)有同源複合結構做參考
如果已經報道了同源蛋白的結構,或者結構功能相似的同類複合結構,則可以通過結構拟合先把兩個待對接的單體分子與參考複合結構疊加,然後撤掉參考結構。這時兩個對接分子的空間位置,就是一個很好的起始位置,以此進行ZDOCK-RosettaDock對接,或者推測出限制性殘基資訊進行HADDOCK對接,都能夠得到比較精确的預測結果。
HADDOCK(https://wenmr.science.uu.nl/haddock2.4/)是一款非常優秀的軟體,使用前需注冊。該軟體需提供兩個待對接分子以及限制性殘基的相關資訊,伯小遠在加入Hd3a和14-3-3c的限制殘基資訊後,獲得的模拟二聚體與真實的晶體結構重疊性極好(這裡圖檔不再展示)。另外,該軟體允許修改大量的參數,靈活性極高,輸出内容豐富而精美,軟體也可用于實際研究。(網站上提供了使用說明,對蛋白質-蛋白質對接有興趣或者有需求的同學,值得好好研究一番)。
3. 對接結果的互作分析
兩個蛋白質能夠互相結合,主要取決于它們之間的靜電引力和範德華力,具體包括:互作界面的電荷分布、幾何形狀互補面積、氫鍵、鹽橋、疏水互相作用、芳環堆積作用等。蛋白質對接的實體、化學原理,模型評價方法是十分複雜的,本文先不讨論。伯小遠僅針對對接結果,提出一般的評估方法。
重點推薦PDBePISA(https://www.ebi.ac.uk/msd-srv/prot_int/pistart.html),該軟體在分析大分子溶劑可及性和互相作用界面方面非常優秀,其可通過輸入複合結構的RSCP PDB編号或上傳對接的複合結構來進行分析,使用方法十分簡單,但面對預測結果,又該如何了解呢?
首先,資訊彙總欄的各項重點名額已在下圖示出,有些參數越大越好,有些越小越好。但沒有明确的能判斷是否互作的标準,是以,最好采用同源蛋白或同類蛋白複合物的結構做為參考;其次,氫鍵、鹽橋的數目越多越好,距離越短越好,也沒有參考門檻值;最後,PDBePISA有一個參數叫複合結構顯著性分數(CSS),它并不是訓示互作可信度的,而是指該界面在複合體形成中的重要程度,即使CSS=0,也不能代表兩分子間沒有互作,但若CSS>0,則兩分子間極可能存在互作。
圖11 PDBePISA對Hd3a/14-3-3c互作界面的分析結果。
三元複合體的組裝
上面一系列的操作獲得了Hd3a/14-3-3c的二進制結構(圖檔未展示),而三元結構是兩個Hd3a/14-3-3c二進制結構依靠14-3-3c和14-3-3c互作聯系起來的,是以還需要建構14-3-3c的同二聚體結構。14-3-3c同二聚體沒有限制性殘基資訊,但其模組化用的高同源模闆(煙草14-3-3c),有同二聚體晶體結構,是以可利用該同源複合結構進行起始位置的初猜,經過ZDOCK分析,模拟的14-3-3c同二聚體複合結構與報道也基本一緻(圖檔未展示)。
然後,就是Hd3a/14-3-3c、14-3-3c/14-3-3c兩個二進制結構的疊加,用SPDBV載入兩個結構,選中全部原子,執行“Fit”菜單的“Magic Fit”,可以快速進行結構疊加(或者叫拟合),經過反複拟合結果展示如下(圖12),與文章報道幾乎一模一樣。
圖12 模拟獲得的Hd3a/14-3-3c複合結構。
接下來,到了最關鍵的OsFD1的疊加。這一步非常困難,由于OsFD1的結構是不準的,報道中的晶體僅包含了OsFD1 189-195的7個殘基,也無法與AlphaFold2的預測結果拟合,疊加的方法完全行不通。是以,隻能依靠酵母雙雜的限制性資訊,使用HADDOCK強行對接。最終全部疊加的結果如下圖,Hd3a/14-3-3c部分與真實結構基本一緻,OsFD1部分與文中的推測有較大差異。注:Taoka等也是靠推測得到的結果,他們的OsFD1是用小鼠的同類蛋白疊加的,可能也不是真實結構喲。
圖13 分子對接和疊加獲得的最終結構。
至此,“劇情”已經不能完全按“編劇”的設想發展了,這場“愛情故事”到此結束!
僅僅依靠模拟就走到這種程度已經非常不容易了,當然需要承認的是,伯小遠在這裡所選的目标蛋白都有不錯的高同源模闆,并且有相關的實驗結果做指導。如果沒有這些,預測的方法是有很大局限性的。為便于大家嘗試,本文提供的方法大多是線上版本的,若需用于研究,建議一定要采用本地版,靈活調整參數,不斷篩選、重複,并配合一定實驗驗證,才能取得更為可靠的結果。
小 結
最後,請大家思考一個問題,為什麼要做那麼多不同的實驗來驗證蛋白間的互作呢?估計有的同學會回答,可以讓論文内容看起來更豐富,容易發表。我隻能說,你真是個小機靈鬼!
如果認真看過我們公衆号以前的文章,可能還記得,我們曾比較過不同的蛋白互作驗證技術間的優缺點,如果隻通過一、兩種驗證方式,可能無法了解事實的真相,也可能得到相悖的結果,這是完全正常的。在Taoka等的研究中,也出現了酵母雙雜的結果與GST pull-down及核磁共振化學位移結果不一緻的情況,這主要是由于OsFD1的192号氨基酸磷酸化導緻的,GST pull-down及核磁共振是使用原核表達的高純蛋白,沒有磷酸化修飾,而酵母内是有磷酸化修飾的。但作者同時采用BiFC對其結果進行驗證,表明OsFD1與Hd3a可能存在互作關系。
酵母雙雜、BiFC、FERT 、Co-IP、GST pull-down、核磁共振,包括本文未涉及的其他技術,如蛋白-蛋白互相作用陷阱、生物膜幹涉等技術,都會因胞内、胞外,有無蛋白質修飾,亞細胞定位是否相同,是否有第三方參與,互作強弱等問題産生差異,解決的辦法就是設定嚴謹的對照、采用盡可能多的方法進行驗證。至于已經忘得一幹二淨的同學,非常有必要再回顧一下《基因功能研究的那些套路你知道多少?(下)》。
References:
protein docking. Nucleic Acids Res. 2008, 36(Web Server issue):W233-8.
Purwestri YA, Ogaki Y, Tamaki S, et al. The 14-3-3 protein GF14c acts as a negative regulator of flowering in rice by interacting with the florigen Hd3a. Plant Cell Physiol. 2009, 50(3):429-38.
Taoka K, Ohki I, Tsuji H, et al. 14-3-3 proteins act as intracellular receptors for rice Hd3a florigen. Nature. 2011, 476(7360): 332-5.
Wigge PA, Kim MC, Jaeger KE, et al. Integration of spatial and temporal information during floral induction in Arabidopsis. Science. 2005, 309(5737): 1056-9.