仅供医学专业人士阅读参考
“生孩子没屁眼”是诅咒成真吗?
孩子出生后,接生人员会按照程序检查新生儿的身体情况,看看孩子身体器官是否完整无畸形,却骤然发现有的孩子天生没有肛门。
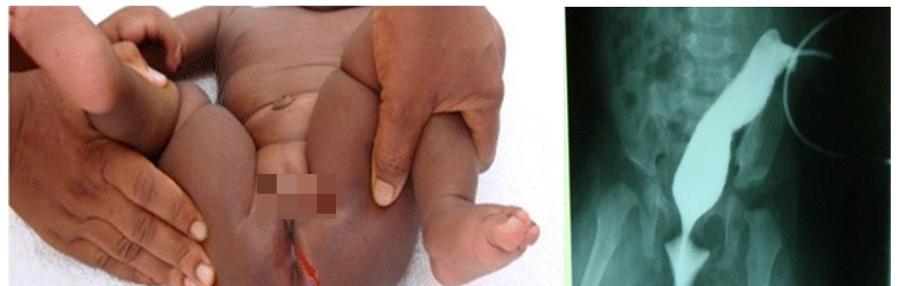
肛门闭锁,图源:文献[1]
民间流传一种说法,缺德事做多后代就会出现无肛儿,这其实是毫无依据的,医学上称这种新生儿缺陷为“肛门闭锁”,是先天性直肠肛门发育畸形的一种临床表现。
一、这是一种常见的消化道畸形
先天性直肠肛门发育畸形其实属于常见的新生儿缺陷疾病,发病率在新生儿中为1:5000-1:1500,占消化道畸形的第一位,一般男婴较女婴多。
除了遗传因素外,父母在产前接触尼古丁、酒精、咖啡因、非法药物、职业危害、超重、肥胖和糖尿病也是其诱发因素。由于没有肛门,宝宝肚子里的胎便无法排出,容易引起肠穿孔、肠梗阻、腹膜炎等并发症,甚至导致死亡。
▌那这种畸形是如何形成的呢?
胚胎早期,婴儿的肛门和直肠尚未分开,与膀胱一同形成泄殖腔。在胚胎发育到第四周左右时,泄殖腔开始出现不同功能器官的分隔,背侧部形成直肠,腹侧部为尿生殖窦,其间为尿直肠,等到胚胎第八周时,才形成正常的直肠和肛管。
如果肛膜未破或肛管发育异常,直肠就无法和外界联通,形成单纯的直肠肛门闭锁。如果在这期间,泄殖腔尿生殖隔发育不全,未能将直肠与尿生殖窦完全分隔,则导致直肠与尿生殖系之间各种瘘管(两个有腔器官之间的病理性排脓管道)的形成。

图源《组织学与胚胎学 (2007年人民卫生出版社)》
一般来说,合并膀胱瘘的几乎都是男孩,女孩的直肠肛门瘘多为阴道瘘。此时患儿虽能通过阴道或尿道排气排便,但粪便留存在尿道或阴道中,容易引起膀胱炎、肾盂肾炎、阴道炎等。
根据临床特点,我们可以将先天性直肠肛门发育畸形分为以下几类:
表1.先天性直肠肛门发育畸形的分类
在先天性直肠肛门畸形病例中,约40%还伴有其他器官发育畸形,相关畸形主要包括泌尿生殖系统(21%-61% 及以上)、脊柱和脊髓(5%-40%)、胃肠道其余部分(10%-25%)和心脏(9%-20%)。特别是泌尿系统的伴随畸形较为多发,在术前应仔细做全身检查。
二、排便异常是最主要的症状
先天性直肠肛门发育畸形的患儿可以首先从外观判断,可发现患儿无肛门或肛门位置异常,或外口狭小。其次还可结合患儿的临床表现,主要包括消化系统的异常。
肛门完全闭锁者,出生后即有急性梗阻表现,出现呕吐,呕吐物含有胆汁或胎粪样物,生后24小时不排便。
若为直肠肛门狭窄或合并其他瘘者,在生后几周、数月或数年出现排便困难、便条很细、腹胀,在左下腹可触及大团粪块,继而出现慢性肠梗阻症状。
从尿道口排气和排粪是直肠泌尿系瘘的主要症状。其中,膀胱瘘因胎粪进入膀胱与尿混合,患儿在排尿的过程中呈绿色尿,但仅在排尿开始时排出少量胎粪,而后胎粪不与尿混合,尿液则是透明的。患儿排尿时可见胎粪和气体排出增多,不排尿时,由于膀胱括约肌的控制,无气体排出。
还可以使用以下辅助检查手段帮助确诊:
1.用最小号金属尿管或探针沿着尿道后壁缓缓插入,通过瘘管进入直肠,用手指触摸肛门部可触及探针尖端,大致明确瘘管的走形、长度和宽度,可以粗略计算直肠盲管与皮肤的距离。
2.X线片显示膀胱内有气体或液体平面时可以确诊为直肠膀胱瘘。直肠肛门狭窄为罕见畸形,瘘管造影可以明确诊断。
三、手术重造肛门
肛门发育畸形主要采取手术治疗,当患儿被确诊后,需根据影像学检查判断出先天性直肠肛门发育畸形患儿直肠末端所处位置,综合手术医生的经验及患儿状态、现有的条件,尽快决定先行结肠造瘘还是直接行根治术。
其中,中高位先天性直肠肛门发育畸形的患者如果无一期手术条件,需先行结肠造瘘,通俗地说就是,把上段的肠子从肚皮上重新开个口,拉出来,制造一个新的排泄口。
肛门闭锁及术后照片(术中发现并闭合了较大的直肠尿道前列腺瘘),图源:文献[2]
结肠造瘘位置及方法的选择至关重要,造瘘口的选择应满足:造瘘口不易脱出或回缩,有利于根治术前造影及清洁远端肠管,不妨碍根治术远端肠管的拖出。因此,临床常选择降结肠或乙状结肠起始部造瘘,乙状结肠起始部因无后腹膜的束缚,好操作,越来越受欢迎。
另外,根据其类型及直肠末端的高度采用不同的根治性手术方法:主要有直肠肛管狭窄扩张法、肛门膜部闭锁切开法、会阴肛门成形法以及直肠瘘管移位肛门成形法。
先天性肛门直肠畸形的治疗效果近年来已有明显改善,总病死率由过去的25%-30%降至近年的10%左右,手术死亡率已降到2%左右。但是目前仍有约1/3的病例术后有不同程度的肛门功能障碍,导致排便异常。
另外,肛门直肠畸形的位置越高,术后排便功能障碍的发生率越高,程度也越严重。而术后排便障碍的发生率,功能障碍高位畸形者为86.4%,中位畸形为47.9%,低位畸形为27%。功能障碍严重者,会给患儿及其家属造成长期的,甚至是终身的痛苦和烦恼,因此除了合理的治疗方法外,坚持术后扩肛和排便训练也十分重要。
参考文献:
[1] Usang UE, Agan TU, Inyang AW, Emehute JD, Itam IH. Syndromic anorectal malformation associated with Holt-Oram syndrome, microcephaly, and bilateral corneal opacity: a case report. J Med Case Rep. 2016 Aug 5;10(1):216.
[2] Pandey A, Gangopadhyay AN, Kumar V, Sharma SP. High anorectal malformation in a five-month-old boy: a case report. J Med Case Rep. 2010 Aug 31;4:296.
[3]夏兴容,刘远梅.中高位先天性肛门直肠畸形治疗进展[J].山东医药,2020,60(10):98-101.
[4] 段泽猛,刘远梅.小儿先天性肛门直肠畸形术后排便功能障碍发生原因与治疗方法的研究进展[J].山东医药,2019,59(23):110-113.
[5] Zwink N, Jenetzky E, Brenner H. Parental risk factors and anorectal malformations: systematic review and meta-analysis. Orphanet J Rare Dis. 2011 May 17;6:25.
[6] 杨红梅,刘琴,孙瑛,贺丽,徐宏宇.随访干预对先天性肛门直肠畸形术后恢复的影响[J].当代护士(中旬刊),2017,(4):57-58.
本文首发:医学界儿科频道
本文作者:史迪仔
责任编辑:CiCi