Non-alternating catalytic copolymerization of intra-chain ketone polyethylene materials
文章出处:Maximilian Baur, Fei Lin, Tobias O. Morgen, Lukas Odenwald, Stefan Mecking. Polyethylene materials with in-chain ketones from nonalternating catalytic copolymerization. Science 2021, 374, 604-607.
Abstract: Polyethylene is the most manufactured plastic in the world, which is composed of inert hydrocarbon chains. The introduction of active polar groups in these chains helps overcome environmental persistence issues and enhances compatibility with other materials. The authors demonstrated that sphosphonophenolic acid coordination Ni complexes can catalyze the non-alternating copolymerization of ethylene and CO, adding low-density intra-chain ketone groups to high molecular weight polyethylene chains while maintaining ideal material properties. After treatment with conventional injection molding techniques, tensile properties remain the same as standard high-density polyethylene, while also being photodegradable.
Polyethylene is the most abundant synthetic polymer, combining convenient processing and low-cost production with beneficial mechanical properties. The latter is caused by the crystallization sequence of stretched hydrocarbon chains. This order is especially pronounced for high-density polyethylene (HDPE), which consists of linear chains with no branches that would otherwise interfere with crystal buildup. Polyethylene is hydrophobic and non-polar and does not easily adhere to metal surfaces or polar materials such as wood. Due to the chemically inert nature of the hydrocarbon chain, polyethylene is not susceptible to degradation reactions and can therefore remain present after being released into the environment.
A promising way to overcome the shortcomings of crystalline polyethylene materials is to catalyze copolymerization with ethylene with a low CO ratio. The ketone groups generated in the polyethylene chain may provide a range of ideal reaction modes, including photodegradability, as demonstrated by branched-chain low-density polyethylene (LDPEs) containing about 1 mol.% keto groups in free radical high-pressure copolymerization. Even in linear polyethylene, low concentrations of ketones are less likely to disrupt crystal order.
Despite the commitment of materials scientists to develop methods of copolymerization, this material remains elusive. In contrast to copolymerization, the selective post-polymerization oxidation of polyethylene requires additional synthetic steps and is not selective for ketone functional groups; it also requires problematic reagents. In copolymerization reactions, the binding of CO to the catalyst is much stronger than that of ethylene monomers, which prevents the continuous binding of olefins. As a result, instead of a mildly perturbed polyethylene chain is formed, alternating polyetones, which are used in high melting point engineering thermoplastics with completely different application and processing properties from polyethylene.
Here, the authors report that the Ni(II) catalyst can enable non-alternating catalytic copolymerization of ethylene and CO. The resulting material has an ideal low content of ketone groups in high molecular weight polyethylene chains, can be melted and processed, and has tensile properties similar to commercial HDPE.
For catalyst selection and design, traditional electron-deficient d0-metal sites, such as those used in polyolefin production, are excluded because they are inactivated by polar molecules such as CO. Quenching such olefin polymerization catalysts with CO is a well-established passivation method. Catalytic processes involving CO, such as olefin carbonylation, typically rely on d8-metal centers, and the alternating polyketones described above are commercially produced with cationic Pd catalysts. In addition, CO, as a ligand widely used in organometallic chemistry, must have a strong binding affinity that must be controlled. Irreversible substitution of other necessary ligands that disrupt the active site must be prevented. In addition, the coordination of CO or keto groups in the growth chain may clog the coordination sites required for further chain growth. It is important to note that the kinetic preference for CO incorporation to promote alternating polyketone formation needs to be overcome. In this sense, the target CO copolymer is opposite to the catalytic copolymerization of ethylene and polar vinyl copolymerization (e.g., acrylates), in which the reactivity of comonomers may be very low.
These arguments prompt consideration of neutral post-transition metal catalysts with strongly bound chelating ligands as promising candidate catalysts. Compared with the more common cationic polymerization catalysts, the preference of the neutral active site for CO binding to other ligands, including monomeric olefins, is not obvious. In fact, the only precedent for the continuous insertion of ethylene in CO copolymerization reactions was observed in neutral phosphine sulfonated Pd catalysts. Despite extensive studies by different joint industrial and academic companies, the reaction resulted in a low molecular weight brittle wax (and possibly heterogeneous composition) with an average molecular weight of Mn ≤ 3000 g·mol-1 and no polyethylene plastic.
Neutral N,O-coordinated dimethyl salicylate and P,O- co-located dimethyl(II) phosphonophenolate Ni(II) catalysts can prepare linear semi-crystalline polyethylene with molecular weight of several million g·mol-1 in ethylene homopolymerization reactions. High polymerization temperatures are also beneficial for the growth of non-alternating chains. For two competing reaction pathways, in the case of different energy barriers (ΔΔG≠), according to ka/kb ~ exp(-ΔΔG≠/RT), the preference for the dynamically dominant reaction pathway decreases as the temperature increases. R is the general gas constant and T is the temperature. The stability of the P,O coordination Ni(II) catalyst is advantageous in this regard. They are suitable for industrial ethylene copolymerization reactions at 140 oC. However, due to its tendency to β-hydride elimination chain transfer pathway, conventional P,O coordination catalysts can only produce oligomers or polymers with molecular weight Mn < 104 g·mol-1 even at lower reaction temperatures. Only recently has this long-standing view been corrected, as it was found that appropriate bulky substituents can strongly inhibit chain transfer, providing high molecular weight polyethylene.
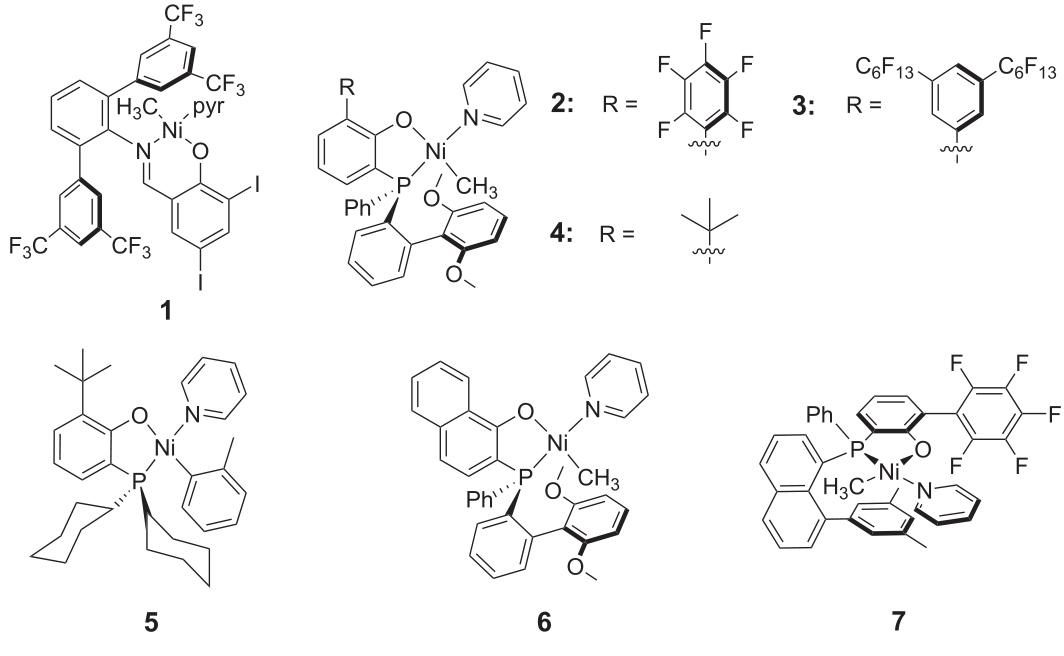
Figure 1
Although the literature agrees that this catalyst is rapidly inactivated by CO and at most only lowly active alternating polyketones are formed, the authors demonstrate that the most advanced salicylaldehyde diimide (Compound 1) and phosphinodiimide (Compound 2) (Figure 1) catalyst precursors, at low CO partial pressure (Table 1), found that ethylene polymerization of these catalysts is optimal at polymerization temperatures. Using compound 1, even if the E / CO ratio > 102, only a small amount of polymer can be obtained. Infrared spectroscopy indicates that the polymer is alternating polyketones. In contrast, a large number of polymers (up to 4 g versus 60 mg when using Compound 1) can be obtained using Compound 2, where the wavelengths of the carbonyl band and alternating polyketones are significantly different (Table 1 and Figure 2B).

Figure 2
Encouraged by these findings, the authors explored a series of custom-modified, state-of-the-art neutral P,O coordination catalyst precursors whose phenolic and aromatic groups cover one or both apical positions (Figure 1). Catalyst precursors (compounds 2-6) are active in the presence of CO and, according to infrared spectroscopy, at high E/CO ratios (E/CO ratio = 100), non-alternating polyketone polymers are formed (Table 1).
Table 1
The microstructure of the polymer was identified by the 13C nuclear magnetic resonance spectroscopy (NMR) method. To improve sensitivity, copolymers obtained from 13CO pressure reactor experiments were used. By combining 1D and 2D NMR methods and referencing data, 1H and 13C NMR spectra can be adequately assigned. This analysis reveals the presence of separated ketones in the polyethylene chain (Figure 2A). Further, the non-alternating order of ketones in adjacent locations can be distinguished, and the alternating motif can be observed. At an ideal CO incorporation value of about 1%, most keto groups are non-alternating, and isolated ketones are ubiquitous. The total ketone content was determined by MRI quantitative analysis and confirmed by infrared spectroscopy (Table 1, Figure 2B). As expected, these in-chain keto group sequences do not affect the ideal material properties of polyethylene. The wide-angle X-ray scattering (WAXS) diffraction pattern of these materials is almost identical to that of HDPE (Figure 3A), emphasizing a conservative solid-state structure. In addition, the melting point and crystallization point have barely changed (Figure 3C). Most notably, ketone-modified polyethylene has a higher molecular weight [mean molecular weight (Mw) of about 400,000 g·mol-1, Mn about 220,000 g·mol-1]. This result is noteworthy because the very low molecular weight observed in ethylene-CO copolymerization produces any partial non-alternating primitives, which is a key factor in useful mechanical properties. In fact, tensile tests on injection molding test rods showed ductility behavior comparable to commercial HDPE (Figure 3D): material elastic modulus E = 1062 ± 53 MPa, tensile strength σy = 26.7 ± 0.2 MPa; literature values for HDPE E = 900 MPa and σy = 27 MPa; literature values for LDPE E = 240 MPa and σy = 12 MPa. There is no evidence of poor crosslinking that hinders melt processing; ketone-based facilitated crosslinking is an issue in the thermoplastic processing of industrial polyketone resins. The exposure experimental results of the keto-polyethylene film in simulated sunlight confirmed its photodegradability. A membrane floating on a water bath is irradiated, with a light intensity equivalent to about 5 months of natural sunlight in southern Europe, and degradation begins to manifest itself as embrittlement and mechanical disintegration of previously ductile samples, and manifests itself as a observable drop in weight by several percentage points. Infrared spectroscopic analysis of 13CO-labeled samples showed that in these short-term degradation experiments, the ketones were only partially consumed; in addition, the formation of new ketones and ester groups allowed the chain to be further degraded. In contrast, the reference sample for the ketone-free homopolymer remains unchanged.
Figure 3
Under the given initial reaction conditions, the composition of the synthetic polymer does not change much as the reaction time or polymer yield increases (Table 1). A homogeneous material is formed consistently, not a perturbation of microstructure and composition (which is the result of an imaginable change in the relative concentration of the monomer). Copolymer yield and mean activity were reduced compared to ethylene homopolymerization (Table 1). However, this is not because of the rapid inactivation of the catalyst. The catalyst is stable for several hours in copolymerization reactions (Table 1). It is hypothesized that, in addition to CO binding, the reversible coordination binding of the formed ketones slows down chain growth to some extent, which is based on observations that, given the E/CO ratio, the overall capacity increases as the monomer concentration increases (Table 1). To overcome this obstacle, a critical polymerization reaction temperature is required. Therefore, effective copolymerization with compound 2 requires about 100 oC, while homopolymerization can be performed quickly at lower temperatures. As expected, a higher polymerization reaction temperature (> 70 oC) will also reduce the incorporation of CO, which is in fact necessary to achieve invariance (Table 1). This result underscores the critical role of the temperature stability of the catalyst (compounds 2-6), enabling non-alternating copolymerization of ketone-modified polyethylene. In addition, the base phosphonophenolic acid matrix also seems to be particularly suitable for non-alternating copolymerization, and the catalyst (Compound 2) is particularly detailed in terms of yield, microstructure and molecular weight of the polymer produced. In contrast, catalysts (Compound 1) can only produce alternating polyketones even at higher polymerization temperatures. This particular property of the phosphonophenolic acid order may be related to strong metal-phosphonic bonds that counteract irreversible inactivation caused by ligand substitution and reduce CO's preference for ethylene incorporation by increasing the electron density at the metal.
High molecular weight ketone-modified polyethylene shows that the presence and incorporation of CO does not promote any problematic chain transfer reactions. In fact, analysis of polymer end groups shows that only those end groups that are also present in homopolyethylene, i.e., olefin end groups derived from β-hydride transfers of Ni-polymer species. This confirms that there are no unwanted additional chain transfer pathways in the CO copolymerization reaction. Instead, the presence of CO appears to hinder chain transfer and branching, which is the conclusion reached by the higher molecular weight and complete absence of branches of copolymers relative to ethylene homopolymers (Table 1), possibly by CO blocking the coordination sites of β-hydride transfer.
On-chain functionalized polyethylene has ideal material properties for HDPE, which provides prospects for less long-lasting photolysis and oxidation chain decomposition of polyolefins in the environment. The reported ketone content is reasonable because the length of the polyethylene segment between the possible break points synthesized corresponds to the length of the mineralizable alkanes. Beyond this rough consideration, the mechanism and rate of degradation will depend heavily on specific environmental conditions, and in this context, an understanding of the ideal profile of the long-term behavior of non-persistent materials remains to be explored.