參考資料
- 生信技能樹 公衆号文章 TCGA資料下載下傳—TCGAbiolinks包參數詳解
- 生信技能樹 公衆号文章 批量COX回歸生存分析圖,指定挑選lncRNA基因,森林圖,ROC曲線打包給你
- 生信星球 公衆号文章 重磅!TCGA資料分析流程梳理總結
- 生信星球 公衆号文章 TCGA3.R包TCGAbiolinks下載下傳資料
- 生信星球 公衆号文章 TCGA的樣本id裡藏着分組資訊
- 簡書文章 TCGA癌症縮寫、癌症中英文對照
- CDSN文章 通過整理TCGA資料,探索某癌症的癌組織和正常組織的差異基因
- TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages
- TCGAbiolinks包下載下傳TCGA資料進行表達差異分析-乳腺癌案例
代碼
資料下載下傳
BiocManager::install("TCGAbiolinks")
library(TCGAbiolinks)
?GDCquery
x<-getGDCprojects()$project_id
x == "TCGA-COAD"
x == "TCGA-READ"
TCGAbiolinks:::getProjectSummary("TCGA-COAD")
query<-GDCquery(project = "TCGA-COAD",
legacy=F,
experimental.strategy ="RNA-Seq",
data.category = "Transcriptome Profiling",
data.type="Gene Expression Quantification",
workflow.type="HTSeq - Counts")
GDCdownload(query)
複制
這裡遇到的問題是:所有資料都下載下傳下來了,如何将資料整合成表達量矩陣的形式呢?
更新
上面遇到的問題今天找到了解決辦法:可以使用
SummarizedExperiment
包中的函數
assay()
函數将表達矩陣提取出來;
colData()
函數好像是獲得一些樣本資訊
檢視TCGAbiolinks包的幫助文檔
browseVignettes("TCGAbiolinks")
嘗試重複幫助文檔中的第3項内容 Downloading and preparing files for analysis
這篇幫助文檔中用到的例子是TCGA-GBM——Glioblastoma multiforme——多形成性膠質細胞瘤
擷取基因表達矩陣
query_3 <- GDCquery(project = "TCGA-GBM",
data.category = "Gene expression",
data.type = "Gene expression quantification",
platform = "Illumina HiSeq",
file.type = "results",
experimental.strategy = "RNA-Seq",
barcode = barcode,
legacy = TRUE)
GDCdownload(query_3)
dataPrep1<-GDCprepare(query=query_3)
library(SummarizedExperiment)
expdat<-assay(dataPrep1)
dim(expdat)
head(expdat)
複制
備注:file.type 的另外一個參數是 normalized_results
DESeq2基因差異表達分析
library(stringr)
group_list<-ifelse(str_sub(colnames(expdat),14,15) == "01","tumor","normal")
group_list
coldata<-data.frame(row.names = colnames(expdat),
condition=group_list)
coldata
library(DESeq2)
expdat<-round(expdat,0)
dds<-DESeqDataSetFromMatrix(countData = expdat,
colData = coldata,
design = ~ condition)
keep<-rowSums(counts(dds))>=10
dds<-dds[keep,]
dds<-DESeq(dds)
res<-results(dds,contrast = c("condition","tumor","normal"))
resOrdered <- res[order(res$pvalue),]
DEG <- as.data.frame(resOrdered)
DEG <- na.omit(DEG)
logFC_cutoff<-with(DEG,mean(abs(log2FoldChange))+2*sd(abs(log2FoldChange)))
logFC_cutoff
DEG$change<-as.factor(ifelse(DEG$pvalue<0.05&abs(DEG$log2FoldChange)>logFC_cutoff,
ifelse(DEG$log2FoldChange>logFC_cutoff,"UP","DOWN"),
"NOT"))
library(ggplot2)
this_title <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3),
'\nThe number of up gene is ',nrow(DEG[DEG$change =='UP',]) ,
'\nThe number of down gene is ',nrow(DEG[DEG$change =='DOWN',]))
ggplot(data=DEG,aes(x=log2FoldChange,y=-log10(pvalue),color=change))+
geom_point(alpha=0.4,size=1.75)+
labs(x="log2 fold change")+ ylab("-log10 pvalue")+
ggtitle(this_title)+theme_bw(base_size = 10)+
theme(plot.title = element_text(size=15,hjust=0.5))+
scale_color_manual(values=c('blue','black','red'))
複制
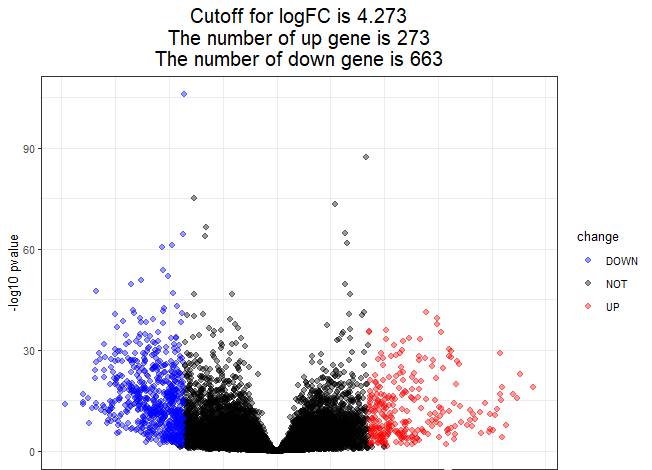
Rplot.png
使用R語言包 clusterProfiler 差異表達基因的GO富集分析
先看一下這個包的幫助文檔
browseVignettes("clusterProfiler")
help(package="clusterProfiler")
library(clusterProfiler)
data(geneList,package="DOSE")
names(geneList)[1:100]
複制
準備自己的輸入資料
df<-DEG[which(DEG$change=="UP"),]
dim(df)
rownames(df)
gene_list<-str_split(rownames(df),"\\|")
gene_list
class(gene_list)
gene_list<-as.data.frame(matrix(unlist(a),ncol=2,byrow = T))
head(gene_list)
yy<-enrichGO(gene=gene_list$V2,
OrgDb='org.Hs.eg.db',
ont="BP",
pvalueCutoff = 0.01)
dotplot(yy)
複制

Rplot01.png
有時間試着用今天分析的資料準備一下GOplot包的輸入資料。