葉綠體基因組結構保守,包含四部分結構:大單拷貝區、小單拷貝區、兩個反向重複區。葉綠體基因組類的文章通常會計算這四個區域的變異位點。那麼第一步便是從完整的葉綠體基因組的序列中分别将這四個區域提取出來,然後比對計算。
本篇文章記錄提取這四個區域用到的python腳本
第一步:利用葉綠體基因組的fasta檔案得到反向重複區的位置資訊
葉綠體基因組類的文章通常是我們自己做幾個,然後結合已經發表的資料做分析。已經公布在NCBI的葉綠體基因組中通常沒有反向重複區的資訊。這個時候就需要我們自己重新注釋。注釋用到的是線上工具GeSeq
https://chlorobox.mpimp-golm.mpg.de/geseq.html
我這裡用4個蘋果屬的葉綠體基因組做示範,序列号分别是:
- NC_031163
- NC_035625
- NC_035671
- NC_036368
将4個fasta檔案上傳到GeSeq,點選送出就可以了
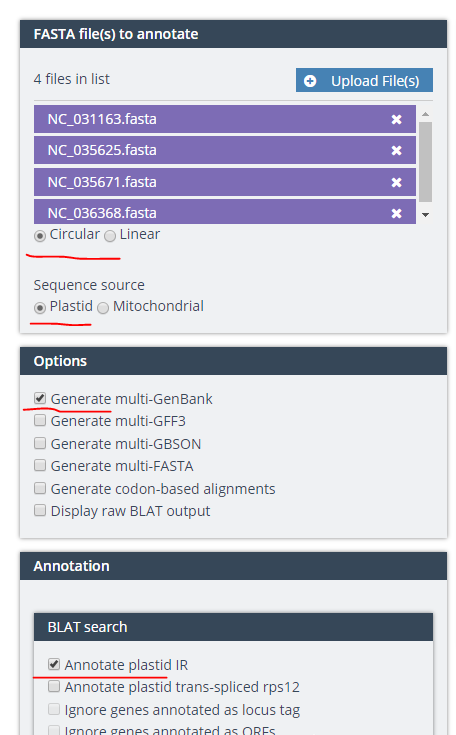
image.png
很快就可以運作完,下載下傳标注的檔案用于後續分析

這個檔案裡包含裡兩個反向重複區的位置資訊
image.png
提取腳本
import os
import sys
from Bio import SeqIO
inputFile = sys.argv[1]
fwLSC = open("LSC_region.fasta",'w')
fwIR = open("IR_region.fasta",'w')
fwSSC = open("SSC_region.fasta",'w')
for rec in SeqIO.parse(inputFile,'gb'):
IR_pos = []
for feature in rec.features:
if feature.type == "repeat_region":
for part in feature.location.parts:
IR_pos.append(part.start)
IR_pos.append(part.end)
if len(rec.seq) == max(IR_pos):
print(rec.id," 可以進行下一步分析!")
IR_pos.sort()
LSC = rec.seq[0:(IR_pos[0]-1)]
IR = rec.seq[(IR_pos[0]-1):IR_pos[1]]
SSC = rec.seq[IR_pos[1]:IR_pos[2]]
fwLSC.write(">%s\n%s\n"%(rec.id,str(LSC)))
fwIR.write(">%s\n%s\n"%(rec.id,str(IR)))
fwSSC.write(">%s\n%s\n"%(rec.id,str(SSC)))
else:
fwLSC.close()
fwIR.close()
fwSSC.close()
os.remove("LSC_region.fasta")
os.remove("IR_region.fasta")
os.remove("SSC_region.fasta")
print(rec.id," 需要調整IR區域的相對位置!")
break
if "LSC_region.fasta" in os.listdir():
print("結果檔案分别是:","LSC_region.fasta ","SSC_region.fasta ","IR_region.fasta ")
else:
print("調整後重新注釋再來提取!")
複制
運作腳本
python .\extract_LSC_SSC_IRs_from_cp_genome.py .\GeSeqJob-20200205-103624_GLOBAL_multi-GenBank.gbff
複制
會提示我
NC_031163. 可以進行下一步分析!
NC_036368. 需要調整IR區域的相對位置!
調整後重新注釋再來提取!
複制
這是因為這條序列的反向重複區位置和通常的不一樣
image.png
因為葉綠體基因組是環狀的,放到檔案裡存儲你可以選擇任意一個堿基作為開始的第一個,葉綠體基因組通常是大單拷貝區的第一個堿基作為起始,但是這條序列不符合普遍情況,我們需要将序列起始的31個堿基放到序列的尾部
用到的腳本
import sys
from Bio import SeqIO
inputFile = sys.argv[1]
pos = int(sys.argv[2])
for rec in SeqIO.parse(inputFile,'fasta'):
fw = open(rec.id+"_1.fasta",'w')
seq_1 = rec.seq[0:pos]
seq_2 = rec.seq[pos:]
fw.write(">%s\n%s\n%s\n"%(rec.id,str(seq_2),str(seq_1)))
fw.close()
複制
使用方法
python .\adjust_IR_pos.py .\NC_036368.fasta 31
複制
然後利用輸出檔案NC_036368.1_1.fasta重新去注釋
注釋完以後再來運作第一個腳本
python .\extract_LSC_SSC_IRs_from_cp_genome.py .\GeSeqJob-20200205-121603_GLOBAL_multi-GenBank.gbff
複制
得到結果
NC_031163. 可以進行下一步分析!
NC_035625. 可以進行下一步分析!
NC_035671. 可以進行下一步分析!
NC_036368. 可以進行下一步分析!
結果檔案分别是: LSC_region.fasta SSC_region.fasta IR_region.fasta
複制
如果需要以上腳本,在我的公衆号留言就可以了!!